Sistemik Lupus Eritematozus Lenfadenopatisi: Olgu Sunumu
Olgumuz 13 yıl önce tanı alan, yaklaşık 1,5 senedir takipsiz olan, hastalık aktivasyonu ve sol boyunda şişlik şikayetiyle aile sağlığı merkezine başvuran 32 yaşında kadın hasta. Fizik muayenede her iki servikal alanda sağda daha belirgin olmak üzere büyüklüğü yaklaşık 2×1 cm, yumuşak kıvamlı, hareketli, ağrısız LAP’lar tespit edildi. Hastadan bu şişliklerin 1,5 aydır devam ettiği, çeşitli antibiyotikler kullanmasına rağmen LAP’larda küçülme olmadığı öğrenildi. Aile sağlığı merkezine başvuran hastanın alınan anamnezi ve yapılan fizik muayenesi sonucu SLE Lenfadenopatisi ön tanısıyla gerekli bilgileri içeren bir sevk notuyla Uşak Eğitim ve Araştırma Hastanesi Genel Cerrahi Kliniği’ne sevk edildi. Daha sonra hastanın yatırılarak tedavi gördüğü hastanedeki ilgili branş hekimleriyle ve hastayla iletişime geçerek yapılan tüm işlemleri takip edildi. Hastaya uygulanan tanısal işlemler ve tedavileri hakkında bilgilenme olanağımız oldu. LAP eksizyon örneğinin histopatolojik tanısı lupus lenfadeniti olarak konuldu. Bu vakalarda erken tanı otoantikorların ve düşük kompleman seviyelerinin saptanamaması nedeniyle zor olabilmektedir. Olgumuzu LAP ile başvuran hastalarda da SLE’nin de ayırıcı tanıda aklımıza gelmesi gereken bir patoloji olduğunu vurgulamak için literatür bilgileri ışığında tartışmayı amaçladık.
Tam Metin
Intoduction
Although the systemic lupus erythematosus (SLE) is seen at every age and in every gender, most of the cases are young female patients in the reproductive age group.(1-9) SLE, which is a multisystemic disease, might be observed to show different clinical findings depending on the organ involvement. In a previous study, the initial symptoms of SLE patients were reported to be fever (67%), arthritis (61%), skin lesions (59%), and lymphadenopathy (LAP) (27.1%), another study reported that the most frequently seen initial symptoms were serositis, polyarthritis, and malar rash.(4,5)
The reactive follicular hyperplasia is a histopathological finding, which is observed in lymph biopsy at most and can be seen at any phase. In the microscopic analysis, the coagulation necrosis and typical hematoxylin particle might not be seen in all cases.(4) It is aimed to discuss the present case together with a literature review in order to draw attention to SLE patients.
Case Presentation
The present case was a 32-year-old female patient, who has been diagnosed 13 years before, had no follow-up for 1.5 years, and presented to the family health center with the complaint of disease activation and swelling on the left side of neck. During her hospitalization period, the results obtained from the full blood test and biochemical analyses are as follows: HDL 27 ↓, WBC 3.53↓, lymphocyte 0.71 ↓, LDH 319 ↑, total protein 8.5 ↑, Fe 48 ↓, and sedimentation ↑. In medical examination, soft, mobile, and painless LAPs, larger of which was 2x1cm, were detected in both cervical regions and the one located on the right side was more remarkable. The body temperature of the patient were normal. Primary and secondary viral markers were analyzed for LAP etiology and no factor was detected.
Hepatitis markers, VDRL, CMV Ig M, EBV Ig M, EBV IgG, EBV VCA IG M, Brucella, Gruber Widal test, and TORCH panel test analysis results were found as negative. The patient stated that the swellings remained there for 1.5 months and the LAPs didn’t reduce although the patient took various antibiotics. In the systemic examination of the patient, no characteristic sign other than photosensitivity in the facial region, weakness, and joint pain was observed.
In neck ultrasonography (USG) images of the patient, many LAPs (the largest one on the right side was 22×9 cm and the largest one on the left side was 15×6 cm) were observed in the bilateral cervical jugular chain and posterior cervical triangle. Since the ANA test because of the leucopenia was positive at 1/300 titer, the anti-dsDNA analysis was performed. Anti-dsDNA and ANCA examinations were also positive. The patient’s existing diagnosis of SLE was confirmed because of the presence of photosensitivity and leucopenia and the positivity of ANA and anti-dsDNA.
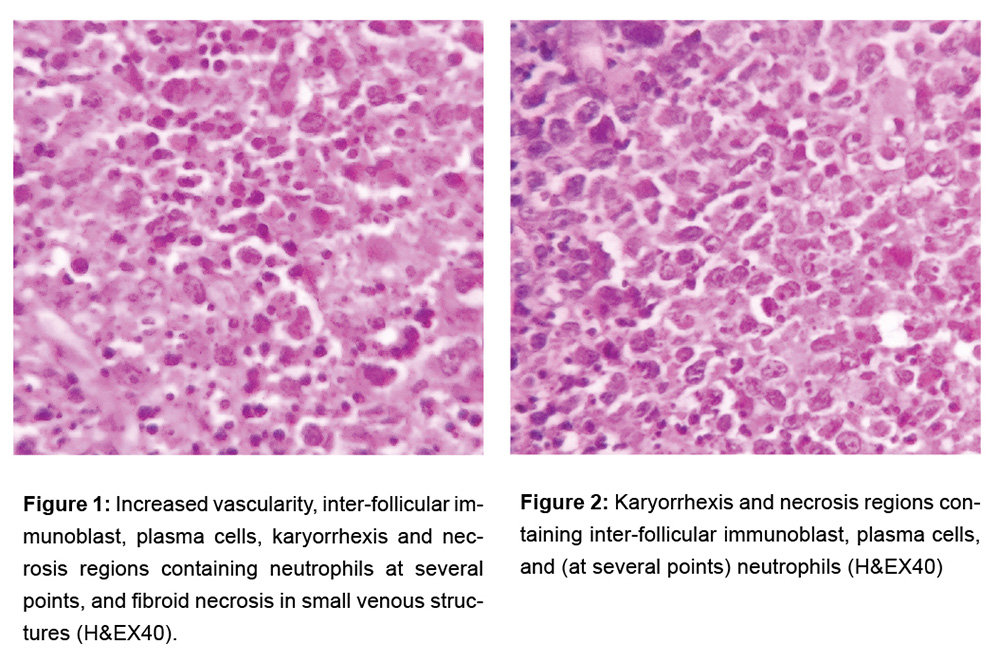
The patient underwent excision for examining the reason for LAP. In the macroscopic analysis, a capsulated off-white/pink lymph node with dimensions of 2.2x2x1.2 cm was detected. In the microscopic analysis, it was observed that the general structure of lymph node was protected but there were increased vascularity, inter-follicular immunoblast, plasma cells, karyorrhexis and necrosis regions containing neutrophils at several points, and fibroid necrosis in small venous structures (Figure 1-2). Based on the current morphology, and clinical and laboratory findings, the case was reported to be lupus lymphadenitis. Patient consent form was received.
Discussion
Although SLE is a multisystemic autoimmune disease that courses with tissue damage arising from the immune system, the beginning with lymph node involvement is very rarely seen.(1,4-6) The etiology is not known exactly. Although it might be seen at any age, it is most frequently seen among female patients aged between 13 and 40 years and the female/male ratio is 9/1. Among the patients applying with complaints of chronic, progressive weakness and loss of appetite symptoms, the immunological diseases (Kikuchi- Fujimoto’s disease) such as SLE should be considered besides the malignity and infectious disorders.(1-5, 10) The present case was a 32-year-old female patient, who presented to the family health center with the complaint of disease activation and swelling on the left side of neck.
The hematological findings are observed in 50-75% of patients, Coombs positive anemia in 30%, leucopenia in 20-40%, and thrombocytopenia in 50%. If in the beginning, LAP is observed in 15-69% of cases. Besides that, the frequency of initial symptoms is sorted as fever, arthritis, skin lesions, and LAP, but it was reported in another study that the most frequently seen beginning symptoms are serositis, polyarthritis, and malar rash.(1,4) The lymph node involvement is rarely seen as an initial symptom in SLE and generalized LAP is observed in retrosternal, mesenteric, and retroperitoneal regions.
The present study has been diagnosed with SLE before and her last application to the clinic was because of cervical LAP complaint. Among the SLE patients, the lymph node involvement is most frequently seen in cervical and axillary regions(2,5) and the involved lymph nodes might be soft and at different sizes, painless or painful, and mobile or fluctuating.(2) It doesn’t extend to deep tissues.(2,5,7) LAP seen in SLE was identified as a benign finding. Benign LAPs are presented by reactive hyperplasia, suppurative lymphadenitis, and granulomatous lymphadenitis.(1,2,5)
The cytological appearance in reactive hyperplasia is polymorphic and it involves a high number of cells. In suppurative lymphadenitis, there are lymphocytes and neutrophils. Epitheloid cell clusters are observed in granulomatous lymphadenitis. The multinuclear foreign bodies, Langhans giant cells, and necrotic material might be seen.(5) In SLE, however, morphologically reactive follicular hyperplasia is seen and infectious and lymphoproliferative disorders can be eliminated in most cases by using lymph node biopsy.(2,5) Lupus lymphadenitis is divided into two types as localized (chains with up to two lymph nodes) and generalized (three or more lymph nodes). Malar erythema, photosensitivity, alopecia, oral ulcer, fever, weight loss, night sweating, and hepatosplenomegaly are the findings observed in SLE patients.(2,5)
From the histopathological aspects, irregular follicles, increased size, hyperplastic germinal center, and cytolysis are observed in the lymph node. Plasmocytosis and interfollicular vascular proliferation histopathologically similar to Castleman’s disease might sometimes be seen and it might cause problem in diagnosis. Atypical paracortical hyperplasia might also be seen in lymphoid follicles and, although it reminds an angioblastic T-cell lymphoma, these two entities are distinguished immunohistochemically (2). Another pathology in the differential diagnosis is Kikuchi-Fujimoto disease (KFD) (histiocytic necrotizing lymphadenitis). This disease is a disorder that involves especially the cervical lymph nodes, affects young patients, and limits itself. Although it has a histopathologically characteristic pattern, it may develop before, together, or after the SLE diagnosis.(1,7-9,11)
KFD is histopathologically characterized with widespread karyorrhexis and patch-like focal necrotic foci accompanied by nuclear wastes and histiocytosis, immunoblasts, and (at several locations) plasma cells and T-lymphocytes. Although the histopathology of SLE includes widespread necrosis and karyorrhexis, it was reported that it can be distinguished from KFD with the presence of hematoxylineosin particles and intense amount of plasma cells especially in the paracortical region, as well as vascular fibrinoid necrosis.(3,2-9,11)
Moreover, whereas there is a low number of cytotoxic T-cell in lupus lymphadenitis, the level of cytotoxic T-cell in Kikuchi-Fujimoto disease is very high. Immunohistochemical staining can be useful.(1,2) In KFD, especially the CD8+ T-lymphocytes are more dominant than CD4+ T-lymphocytes.(9) Another group that should be considered in the differential diagnosis is the diseases related with IgG4. In this group of diseases, the tissue infiltration consisting of IgG4 positive plasma cells and T lymphocytes is accompanied by fibrosis and the level of serum IgG4 is high.(1,5,9)
The immunopathogenesis of SLE is related with the loss of self-tolerance determined genetically and with cellular activation developing based on the non-genetic factors such as environmental, hormonal, and infectious factors. The activation of T lymphocytes with γ-interferon triggers the progressive and permanent dilatation of polyclonal B lymphocytes that are resistant to apoptosis and creating the auto-antibody features of disease.(2) In some of the previous studies, it was reported that the malignity risk increased in autoimmune diseases such as SLE.(1,2) The re-evaluation of diagnosis is very important especially for the patients with treatment-resistant SLE. The risk of lymphoma, especially the risk of Non-Hodgkin’s lymphoma, significantly increases in SLE patients. The lymph node biopsy can provide very useful information at this stage. Immunohistochemical staining would be very useful in the differential diagnosis.(2)
As a result, it is seen in our case that a disease such as SLE lymphadenitis, normally diagnosed and performed follow-ups in 2nd or 3rd level health institution, can be diagnosed in a primary care health institution by family physicians who have a careful eye and holistic approach.With this case report, it has been shown that systemic and chronic diseases can be diagnosed in the early stages of the disease with careful medical history taking and physical examination.
Referanslar
- Albayrak İ, Küçük A, Bağçacı S, Küçükşen S, Tunç R. Lenfadenopati ile başlayan sistemik lupus eritematozus: Olgu sunumu. FTR Bil Der 2014; 17: 38-41.
- Neto NSR, Bonfiglioli KR, Milanez FM, Macêdo PA, Levy-Neto M. Lymphadenopathy and systemic lupus erythematosus. Bras J Rheumatol 2010;50 (1) : 96-101.
- Gelincik İ. Bir Nekrotizan Lenfadenit Nedeni: Kikuchi – Fujimoto Hastalığı: Olgu Sunumu. Yeni Tıp Dergisi 2012;29(3):176-8.
- Türkbeyler İH, Pehlivan Y, Çömez G, Zengin O. Göktepe F, Onat AM. Yaygın lenfadenopati nedeni ile başvuran hastada sistemik lupus eritematozis tanısı: Olgu sunumu. ADÜ Tıp Fakültesi Dergisi 2011; 12(2): 45-7.
- Shrestha D, Dhakal AK, Shiva Raj KC et al. Systemic lupus erythematosus and granulomatous lymphadenopathy. BMC Pediatrics 2013;13:179-85.
- Naramala S, Adapa S et al. An Overlapping Case of IgG4 Related Disease and Systemic Lupus Erythematosus. Journal of Investigative Medicine High Impact Case Reports 2019;7: 1–4.
- Aktaş B, Kalyoncu U, Çağkan İA et al. Clinicopathologic case discussion: systemic lupus erythematosus. RAED Dergisi 2014;6(1):36-4.
- Çiçek D, Dağlı AF. Kikuchi – Fujimoto hastalığı ile karışan bir sistemik lupus eritematozus olgusu. Fırat Tıp Dergisi 2007;12(2): 137-9.
- Akhüseyinoğlu M, Saylam G, Han Ü et al. A Rare necrotising lymphadenitis cause in our country: Kikuchi-Fujimoto Disease. KBB ve BBC Dergisi 2009;17 (2):58-61.
- Scott GD, Kumar J, Oak JS, Boyd SD, Raess PW, Gratzinger DA. Histology-Independent Signature Distinguishes Kikuchi-Fujimoto Disease/Systemic Lupus Erythematosus-Associated Lymphadenitis From Benign and Malignant Lymphadenopathies. Am J Clin Pathol 2020;154(2):215-24. doi:10.1093/ajcp/aqaa036.
- Masab M, Surmachevska N, Farooq H. Kikuchi Disease. In: StatPearls. Treasure Island (FL), StatPearls Publishing; 2020.